Building Gapless Genomes for Conservation
|
In collaboration with Colossal Foundation and the University of Melbourne, we are developing telomere to telomere (T2T) scale genomes and epigenomes for a variety of eukaryotic species. With these data, we can work with conservation management teams to provide genomic resources for conservation management strategies, provide a genetic framework for genetic rescue, and address key questions about genome and chromosome evolution. |
 |
Exploring genomes to understand genetic instability
|
The development of genome-scale sequence data has provided a foundation for studying biological processes in targeted species, while the ability to compare the genomes of different species has afforded a greater understanding of genome evolution and the functional elements dispersed among coding and non-coding regions. We are capitalizing on the advantages in employing comparative genomics approaches to understanding genome and species biology to understand lability in centromeres, telomeres and chromosome breakpoints. Within marsupials, the macropodids (kangaroos and wallabies) exhibit rapid chromosome evolution while a sister group, the dasyurids, are chromosomal stable. We are using comparative genome assemblies to build genetic and epigenetic models to uncover the genomic features that guide stability vs instability. How do specific repeats and epigenetic features mediate genome stability? |
 |
Revealing genomic mechanisms of rapid chromosomal speciation
 |
Through comparative genomics and cytogenetics, we have brought to bear newly developed genome assembly tools on a non-traditional model system that exhibits rapid karyotypic evolution: the lesser apes - gibbons. We aim to address key questions on the role retroelements play in rapid chromosome change, centromere function and speciation. How does the activity of retroelements impact chromosomal speciation? |
Dissecting centromere establishment and assembly in humans
|
Centromeres are the site of kinetochore assembly and spindle attachment to chromosomes during mitosis and meiosis in eukaryotic organisms. The proper functioning of centromeres is essential to the faithful segregation of genetic material in each cell generation, and errors in the cascade of events circumscribing spindle attachment lead to genome instability and aneuploidy. However, major hurdles in understanding centromere evolution and function lie in the highly repetitive nature of most centromeric DNA and an inability to decouple centromere divergence from species evolution and stochastic processes such as genetic drift and molecular drive. We have developed genomic and cell engineering tools to study the genomic and epigenomic features involved in centromere function in humans. How do specific repeats and epigenetic features demarcate active centromeres? |
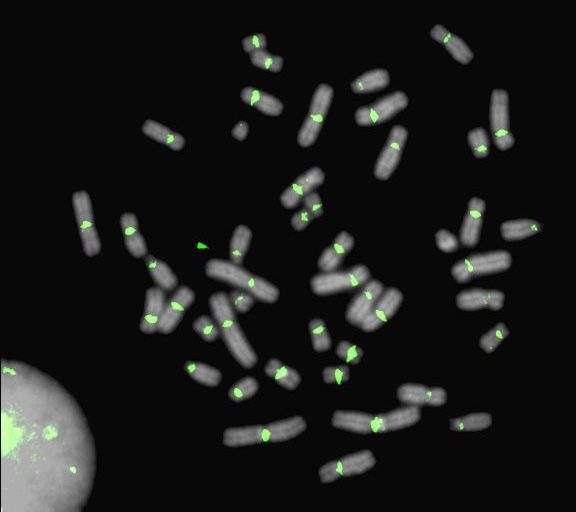 |
Using comparative genomics to understand adaptive responses within marine ecosystems
 |
Despite the advantages in employing comparative genomics approaches to understanding genome and species biology, few species that occupy pivotal roles in marine ecosystems and pelagic food webs have been utilized as model organisms for genome-scale studies. Employing diverse technologies for the development of reference genome assemblies, we have targeted several species for comparative genomics that will afford a greater understanding of adaptive responses to environmental variation and the evolution of novel phenotypes within marine ecosystems. One of our target species is the Southern Ocean salp, Salpa thompsoni, which has shown altered distribution and abundance in Antarctic Ocean ecosystems in recent years. With a high-quality genome assembly, we can begin to unravel the genomic features that allow the salp to adapt to our warming oceans with the devastating consequence of depleting the ocean of the extraordinary diversity of organisms that rely on krill. |
Developing genome resources and novel sequencing applications
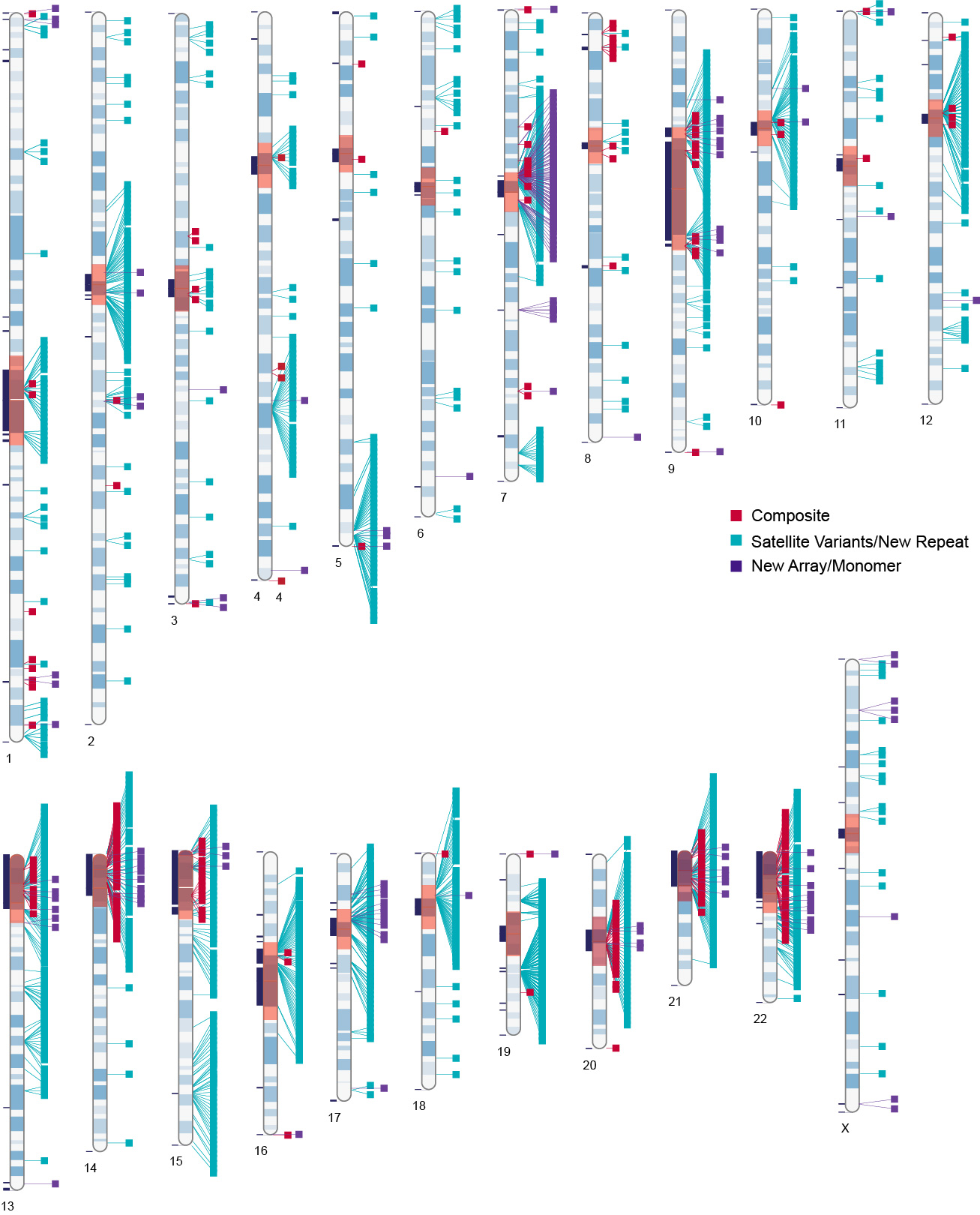 |
Rapidly evolving sequencing technology has enabled more comprehensive, genome-scale analyses of genome structure and function, gene expression networks, and population diversity. Coincident with this advance is the capacity to establish broad eukaryotic species as models for studying genome biology. My lab has a long-standing interest in developing new model systems with broad applications to scientists as well as novel methodologies for assessing genome sequences. To this end, we have worked on genome projects for over 40 different species, have developed computational tools to merge various forms of data for scaffolding, including non-sequence based data, and have established workflows for using these genome assemblies for understanding the dynamics of gene expression and epigenetics in various contexts. Part of our efforts have focused on understanding the transcriptional landscape of transposable elements in centromeres of various model species, including human. |