Web cookies (also called HTTP cookies, browser cookies, or simply cookies) are small pieces of data that websites store on your device (computer, phone, etc.) through your web browser. They are used to remember information about you and your interactions with the site.
Purpose of Cookies:
Session Management:
Keeping you logged in
Remembering items in a shopping cart
Saving language or theme preferences
Personalization:
Tailoring content or ads based on your previous activity
Tracking & Analytics:
Monitoring browsing behavior for analytics or marketing purposes
Types of Cookies:
Session Cookies:
Temporary; deleted when you close your browser
Used for things like keeping you logged in during a single session
Persistent Cookies:
Stored on your device until they expire or are manually deleted
Used for remembering login credentials, settings, etc.
First-Party Cookies:
Set by the website you're visiting directly
Third-Party Cookies:
Set by other domains (usually advertisers) embedded in the website
Commonly used for tracking across multiple sites
Authentication cookies are a special type of web cookie used to identify and verify a user after they log in to a website or web application.
What They Do:
Once you log in to a site, the server creates an authentication cookie and sends it to your browser. This cookie:
Proves to the website that you're logged in
Prevents you from having to log in again on every page you visit
Can persist across sessions if you select "Remember me"
What's Inside an Authentication Cookie?
Typically, it contains:
A unique session ID (not your actual password)
Optional metadata (e.g., expiration time, security flags)
Analytics cookies are cookies used to collect data about how visitors interact with a website. Their primary purpose is to help website owners understand and improve user experience by analyzing things like:
How users navigate the site
Which pages are most/least visited
How long users stay on each page
What device, browser, or location the user is from
What They Track:
Some examples of data analytics cookies may collect:
Page views and time spent on pages
Click paths (how users move from page to page)
Bounce rate (users who leave without interacting)
User demographics (location, language, device)
Referring websites (how users arrived at the site)
Here’s how you can disable cookies in common browsers:
1. Google Chrome
Open Chrome and click the three vertical dots in the top-right corner.
Go to Settings > Privacy and security > Cookies and other site data.
Choose your preferred option:
Block all cookies (not recommended, can break most websites).
Block third-party cookies (can block ads and tracking cookies).
2. Mozilla Firefox
Open Firefox and click the three horizontal lines in the top-right corner.
Go to Settings > Privacy & Security.
Under the Enhanced Tracking Protection section, choose Strict to block most cookies or Custom to manually choose which cookies to block.
3. Safari
Open Safari and click Safari in the top-left corner of the screen.
Go to Preferences > Privacy.
Check Block all cookies to stop all cookies, or select options to block third-party cookies.
4. Microsoft Edge
Open Edge and click the three horizontal dots in the top-right corner.
Go to Settings > Privacy, search, and services > Cookies and site permissions.
Select your cookie settings from there, including blocking all cookies or blocking third-party cookies.
5. On Mobile (iOS/Android)
For Safari on iOS: Go to Settings > Safari > Privacy & Security > Block All Cookies.
For Chrome on Android: Open the app, tap the three dots, go to Settings > Privacy and security > Cookies.
Be Aware:
Disabling cookies can make your online experience more difficult. Some websites may not load properly, or you may be logged out frequently. Also, certain features may not work as expected.
Exploring genomes to understand genetic instability
The development of genome-scale sequence data has provided a foundation for studying biological processes in targeted species, while the ability to compare the genomes of different species has afforded a greater understanding of genome evolution and the functional elements dispersed among coding and non-coding regions. We are capitalizing on the advantages in employing comparative genomics approaches to understanding genome and species biology to understand lability in centromeres, telomeres and chromosome breakpoints. Within marsupials, the macropodids (kangaroos and wallabies) exhibit rapid chromosome evolution while a sister group, the dasyurids, are chromosomal stable. We are using comparative genome assemblies to build genetic and epigenetic models to uncover the genomic features that guide stability vs instability.
How do specific repeats and epigenetic features mediate genome stability?
Revealing genomic mechanisms of rapid chromosomal speciation
Through comparative genomics and cytogenetics, we have brought to bear newly developed genome assembly tools on a non-traditional model system that exhibits rapid karyotypic evolution: the lesser apes - gibbons. We aim to address key questions on the role retroelements play in rapid chromosome change, centromere function and speciation.
How does the activity of retroelements impact chromosomal speciation?
Dissecting centromere establishment and assembly in humans
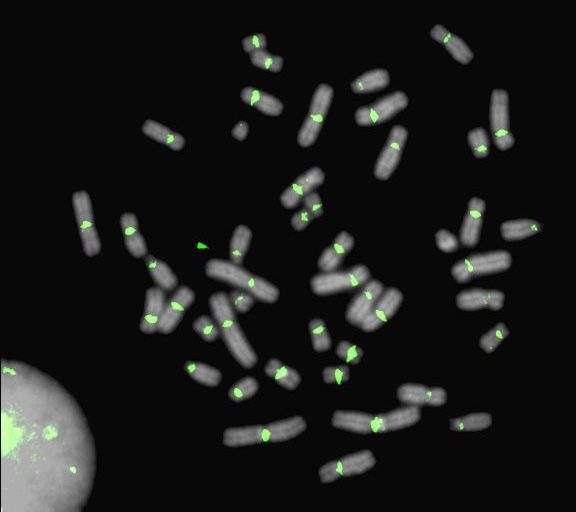
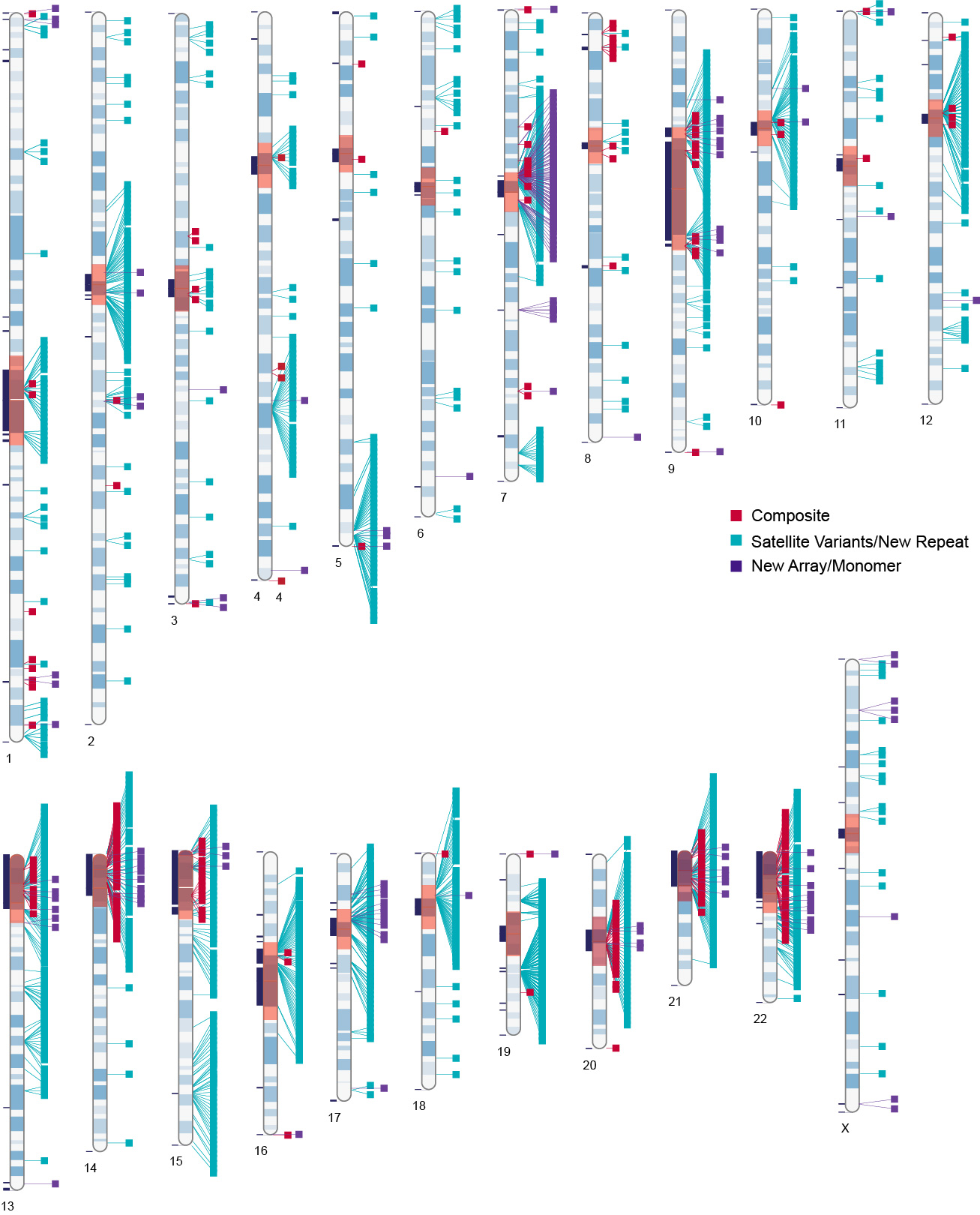
Centromeres are the site of kinetochore assembly and spindle attachment to chromosomes during mitosis and meiosis in eukaryotic organisms. The proper functioning of centromeres is essential to the faithful segregation of genetic material in each cell generation, and errors in the cascade of events circumscribing spindle attachment lead to genome instability and aneuploidy. However, major hurdles in understanding centromere evolution and function lie in the highly repetitive nature of most centromeric DNA and an inability to decouple centromere divergence from species evolution and stochastic processes such as genetic drift and molecular drive. We have developed genomic and cell engineering tools to study the genomic and epigenomic features involved in centromere function in humans.
How do specific repeats and epigenetic features demarcate active centromeres?
Using comparative genomics to understand adaptive responses within marine ecosystems
Despite the advantages in employing comparative genomics approaches to understanding genome and species biology, few species that occupy pivotal roles in marine ecosystems and pelagic food webs have been utilized as model organisms for genome-scale studies. Employing diverse technologies for the development of reference genome assemblies, we have targeted several species for comparative genomics that will afford a greater understanding of adaptive responses to environmental variation and the evolution of novel phenotypes within marine ecosystems. One of our target species is the Southern Ocean salp, Salpa thompsoni, which has shown altered distribution and abundance in Antarctic Ocean ecosystems in recent years. With a high-quality genome assembly, we can begin to unravel the genomic features that allow the salp to adapt to our warming oceans with the devastating consequence of depleting the ocean of the extraordinary diversity of organisms that rely on krill.
Developing genome resources and novel sequencing applications
Rapidly evolving sequencing technology has enabled more comprehensive, genome-scale analyses of genome structure and function, gene expression networks, and population diversity. Coincident with this advance is the capacity to establish broad eukaryotic species as models for studying genome biology. My lab has a long-standing interest in developing new model systems with broad applications to scientists as well as novel methodologies for assessing genome sequences. To this end, we have worked on genome projects for over 40 different species, have developed computational tools to merge various forms of data for scaffolding, including non-sequence based data, and have established workflows for using these genome assemblies for understanding the dynamics of gene expression and epigenetics in various contexts. Part of our efforts have focused on understanding the transcriptional landscape of transposable elements in centromeres of various model species, including human.
Deep Ocean Genomes Project
Earth’s ocean is the largest and most biodiverse ecosystem on Earth, hosting 33 known phyla from the tree of life, with ~410,000 named species and estimates of >100 million species. The deep sea hosts a broad spectrum of habitats including hydrothermal vents, methane seeps, oxygen minimum zones, seamounts, canyons and trenches with evidence suggesting deep-ocean life is richly diverse and highly adapted. In partnership, Woods Hole Oceanographic Institute (Tim Shank) and the University of Connecticut have established the Deep-Ocean Genomes Project (DOG). We are focused on implementing genomics technologies to address diverse ecological and evolutionary hypotheses within and across a myriad of deep-sea habitats and lineages.
What are the genomic components of evolutionary novelty and environmental adaptation?